使用新开发的珠磨机在甲基丙烯酸甲酯(MMA)中制备分散良好的 TiO2(二氧化钛)纳米颗粒悬浮液,并通过后续的 TiO2-MMA 悬浮液聚合合成 TiO2- PMMA 纳米复合材料。在TiO2- MMA 悬浮液中加入偶联剂(3-丙烯氧基丙基)三甲氧基硅烷(APTMOS)后,珠磨机成功地分散了二氧化钛纳米颗粒团聚体。在纳米颗粒质量分数高达 0.05 的悬浮液中,团聚颗粒被破碎成小至 10 nm 的初级颗粒。分散良好的二氧化钛纳米颗粒悬浮液降低了紫外线透射率,但可见光透射率与纯 MMA 相似。TEM 图像显示,经珠磨机研磨后的纳米粒子在二氧化钛-PMMA 纳米复合材料中保持良好分散,并且向 PMMA 中添加二氧化钛纳米粒子提高了 PMMA 的热稳定性。旋涂二氧化钛-PMMA薄膜比纯PMMA薄膜具有更高的折射率,其中二氧化钛重量百分比越高的薄膜具有越高的折射率。
1. 简介
纳米大小的粒子对于许多应用都是必不可少的,因为纳米粒子与宏观颗粒相比,具有显著不同的电子、光学、磁性和催化性质。将纳米粒子并入基质材料(即聚合物溶液或熔体或弹性体)中通常是有利的,以产生由填充纳米粒子带来的额外性能的纳米复合材料。最近的研究表明,聚合物内的纳米颗粒填料可以改变聚合物的热力学性质,如弹性模量粘度和玻璃化转变温度,以及光学性质。纳米粒子-聚合物复合材料制备方面的进展将有助于开发新型数据存储、光学和电流变材料。纳米粒子-聚合物复合物可通过在聚合物基质中合成纳米粒子或通过将纳米粒子分散于单体中并在纳米粒子存在下使单体聚合来制备。前一种方法受到以下事实的限制:并非所有类型的纳米粒子都能在聚合物基质内容易合成。由于许多纳米粒子可以通过火焰合成等廉价工艺大量合成 ,因此与在聚合物基质中生产纳米粒子相比,纳米粒子单体分散体的聚合可以用于制备更多种纳米粒子-聚合物复合材料。然而,在液体悬浮液中,纳米粒子之间的吸引力往往足够大 ,因此纳米粒子往往在大多数溶剂中团聚。纳米颗粒在单体溶液中的分散不良不会形成纳米颗粒-聚合物纳米复合物;相反,在聚合后,它们将形成纳米颗粒-聚合物微复合材料,其中大的纳米颗粒团聚被嵌入聚合物基质中。因此,为了制备纳米聚合复合材料,有必要开发分散纳米粒子在单体溶剂中的方法。
机械研磨过程常用于破碎颗粒团聚体并使其分散在悬浮液中,然而,大多数商业化的研磨设备不能完全分解纳米颗粒的团聚体。最近开发的珠磨机被证明在水悬浮液中将超微米尺寸的 TiO2 纳米颗粒团聚体分解成 15 nm 初级颗粒,而不影响颗粒结晶度。这种珠磨机可以使用小至 15 μm 的磨珠,因为它利用离心力将磨珠与纳米颗粒悬浮液分离,使悬浮液污染小。较小的磨珠与纳米粒子的碰撞能量较低,因此可以防止纳米颗粒在研磨过程中受损,同时分散纳米颗粒团聚体。
在本研究中,我们使用离心珠分离的珠磨机来制备稳定的纳米粒子-单体分散体,然后单体聚合形成纳米颗粒聚合物复合材料。该过程的示意图如图 1 的 a 部分所示。据我们所知,这是使用研磨操作将纳米粒子分散到用于聚合物-纳米粒子复合材料合成的单体溶剂中的第一份报告。作为模型体系,TiO2(二氧化钛)纳米粒子分散在甲基丙烯酸甲酯(MMA)单体中。聚甲基丙烯酸甲酯(PMMA)是一种对可见光透明的重要热塑性材料。在 PMMA 中添加 TiO2纳米粒子可以调节 PMMA 的折射率,增强 PMMA 对紫外线的吸收能力。研究了研磨时间、偶联剂的使用和纳米颗粒表面改性对 MMA 中二氧化钛团聚体粒径分布的影响。采用动态光散射、紫外-可见光光谱、热重分析、傅里叶变换红外光谱、光学和电子显微镜等方法评估球磨对TiO2-MMA 分散的影响,以及 TiO2-PMMA 复合材料的性能。
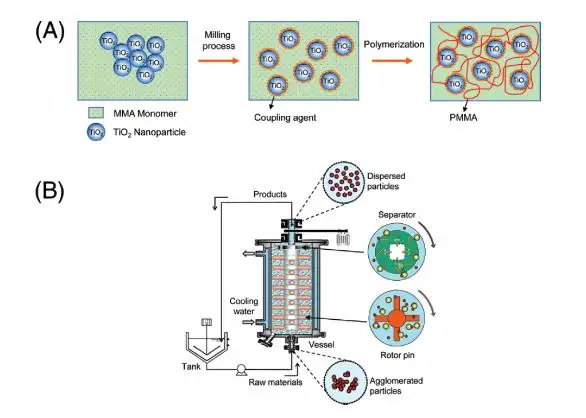
图 1 (a)研磨-聚合过程的表示 (b)采用离心珠分离的珠磨系统示意图
2. 实验部分
材料 两种商业生产的二氧化钛纳米颗粒,标称初始粒径为15 nm的表面未修饰的颗粒(MT150A,金红石相;Tayca Co. Ltd,日本)和初始粒径为 15 nm 的烷基硅烷表面改性粒子(NKT90,锐钛矿相;日本 Aerosil 株式会社)被用于实验。采用透射电子显微镜(TEM,日本电子光学实验室 jm - 3000f,源型 jm -2010, 200 kV)测定表面改性后的粒子初始粒径。两个样品的 x 射线衍射(XRD, Rigaku, RINT2550, VHF)图案表明它们与制造商的测量相相似。偶合剂(3-丙烯氧基丙基)三甲氧基硅烷(APTMOS;KBM5103, Shinetsu Co. Ltd.,日本)也被用于纳米颗粒悬浮液。珠磨含有 15 μm ZrO2(氧化锆)珠(Neturen Co., Ld. Tokyo, Japan)。在制备纳米颗粒悬浮液之前,通过蒸馏纯化 MMA 单体溶剂(三菱气体化学,纯度 99.8%,25℃0.569cp)。采用偶氮双异丁腈(AIBN,大冢化学有限公司纯度:>99%)作为聚合引发剂。
研磨过程 图 1 的 b 部分显示了珠磨机的原理图,对珠磨机的详细描述可在其他地方找到。珠磨机由一个 80ml 容器、泵和混合罐组成。纳米颗粒悬浮液被泵入含有氧化锆珠和离心转子的容器中。磨珠在容器的下部(分散部分)被搅动,这促使聚集的颗粒破碎。悬浮液从分散部分泵入到上部区域(离心部分),离心力用于将氧化锆珠与纳米颗粒悬浮液分离。离心力还可去除大的纳米颗粒聚集体,直至将其破碎成小粒子。然后将纳米粒子悬浮液回收回分散段。
为了防止系统温度升高,容器被安置在冷却水套中,并与外部环境完全密封。 为了破碎悬浮在 MMA 中的二氧化钛附聚物,将二氧化钛质量分数范围为 0.01 至 0.1 的二氧化钛-MMA 悬浮液泵入珠磨机,循环质量流量为 10 kg/h(研磨中的停留时间为 29 秒)区域,每小时 25 个循环,总再循环时间 144 秒)。添加或不添加偶联剂制备纳米粒子悬浮液。通过以 10 m/s 的速度旋转外圆柱壁,产生用于容器上部珠粒分离的离心力。氧化锆珠占据容器体积的65%。
聚合 研磨后,将二氧化钛-MMA悬浮液转移至反应烧瓶中,向其中添加聚合引发剂AIBN使得AIBN与MMA的质量比为0.001。聚合在60℃、N2气氛下进行6小时。
涂片 将二氧化钛- PMMA 纳米复合材料(1g)溶解于玻璃烧瓶(50 mL)中的 20 mL 四氢呋喃中并使用旋转涂布器(1H-D7, Mikasa)将其旋转涂覆到蓝宝石衬底(Kyocera Co., Ltd)上。在 2000 rpm 下进行旋涂以 1 mL 的滴加体积。
材料表征 使用动态光散射 (DLS) 和 FPAR-1000(Otsuka Denshi.Co.,塑料比色皿,1 mL 体积)测量选定研磨时间后的粒度分布。尺寸分布测量无需稀释,一式两份进行,以确保重现性。MMA 的折射率和粘度用作仪器内置软件包的输入参数,用于尺寸分布反卷积。将样品干燥并与 KBr 粉末混合用于 FTIR 测量(PerkinElmer,Spectrum One System A,范围:7800-350 cm-1,分辨率:0.5-64 cm-1,透射模式)。将样品倒入光学深度为 1 cm 的石英池中,并通过紫外可见光谱(U-2810,Hitachi,190-1100 nm)进行测量。使用透射电子显微镜观察 MMA 聚合后的 TiO2 纳米粒子。为了获得 TEM 图像(JEOL-JEM-2010,200kV),使用切片机将 TiO2 -PMMA 复合材料切成薄层,并将单个薄层放置在 TEM 网格上。使用热重分析仪(Shimadzu Thermo Plus TG8120;最高温度,800 °C;升温速率,N2 中 5 °C/min)测量 TiO2 -PMMA 纳米复合材料的热性能。使用棱镜耦合器(型号 2010,Metricon Corp.)在 633 nm 波长下测量旋涂复合膜的折射率。
3. 结果与讨论
MMA 中的 TiO2团聚破裂
为了制备分散良好的纳米颗粒悬浮液,需要优化研磨操作条件以及控制纳米颗粒和纳米颗粒团聚体之间的吸引力和斥力。随着团聚体的破碎,悬浮液中粒子的数量浓度大幅增加,这可能导致粒子重新团聚,特别是在粒子之间存在吸引力的情况下在悬浮液制备前对纳米粒子进行表面改性可防止发生再团聚。首先研究了在没有任何偶联剂的情况下,悬浮在甲基丙烯酸甲酯(MMA)中的未修饰和表面修饰的二氧化钛纳米粒子的粉碎性能。结果表明,纳米颗粒的表面改性对磨珠性能没有影响。最初,二氧化钛纳米颗粒团聚,团聚粒径在超微米范围内。DLS 测量表明,在整个磨矿过程中,大团聚体仍然存在于悬浮液中。在研磨后通过电感耦合等离子体光谱检查了悬浮液的化学成分,发现氧化锆珠没有污染样品(它们在研磨中通过离心从悬浮液中分离出来,因为它们比二氧化钛密度更大),而且大团聚体实际上是二氧化钛。因此,球磨对粒径分布几乎没有影响,可以推测,在珠磨过程中破碎的团聚体会迅速重新团聚。在研磨数小时后,团聚的二氧化钛纳米粒子也从 MMA 单体的悬浮液中沉淀出来。
珠磨机不能同时分散未修饰和表面修饰的二氧化钛纳米粒子,这意味着二氧化钛在 MMA 中相互强烈吸引( Hamaker 常数很大)。硅烷偶联剂通常用于将无机材料粘结到有机材料上 21;因此,在研磨前将微量(对于下文的所有实验,每平方米二氧化钛一次粒子表面积为 2 g 偶联剂)偶联剂 APTMOS 添加到二氧化钛- MMA 悬浮液中。图 2 的 a 部分显示了 5、60、120、180 和 240 分钟的珠磨时间后,纯二氧化钛粉末以及未经改性的 TiO2-MMA 悬浮液的 FTIR光谱。在悬浮 FTIR 光谱中,2840 cm-1 (A,甲氧基官能团的 C- h 拉伸)、1640-1590 cm-1 (B,APTMOS 的 CdC)和 1110-1000 cm-1 (C, Si-O 拉伸(Si-O- r))的峰明显。随着研磨时间的增加,在 1110-1000 cm-1 处的峰强度降低,这是由于 Si-O-R 键中的甲基被钛取代。因此,FTIR 光谱表明,偶联剂在研磨过程中包裹了二氧化钛颗粒。在偶联剂的作用下,微球研磨可以成功地将纳米粒子团块粉碎成与原始粒子大小相同的粒子。
图 2 的 b 和 c 部分分别显示了未经修饰和表面修饰的二氧化钛纳米粒子的 MMA 悬浮液的粒径分布(y 轴,DLS 测量的强度)。首先,在二氧化钛团块暴露的表面涂上偶联剂。随着珠磨的进行,新的二氧化钛表面被暴露出来,然后被偶联剂涂覆,以防止再团聚的发生。与表面改性的二氧化钛(90 min)相比,未改性的二氧化钛(150 min)所需的时间更长。这可能是由于未修饰的二氧化钛纳米颗粒的团聚体比表面修饰的二氧化钛颗粒具有更强的颗粒间力。珠粒和团聚体之间的碰撞产生的能量是珠粒和团聚体速度、珠粒大小和团聚体大小的函数 ,但相对独立于使团聚体聚集在一起的粒子间力。只有一定比例的碰撞才有足够的能量来破坏凝聚体内的粒子间力。在未修饰和表面修饰的二氧化钛中,团聚珠碰撞的能量分布可能是相同的。然而,未经改性的二氧化钛粒子之间的作用力可能比表面改性的二氧化钛粒子之间的作用力更强。较小比例的碰撞随后导致团聚破碎;因此,需要更长的研磨时间来打破未修饰的二氧化钛纳米颗粒的团聚体。因此,减少必要的研磨时间不仅可以通过优化珠磨条件来实现,还可以通过使用纳米颗粒来实现,因为纳米颗粒只能形成弱结合的或软的团聚体。
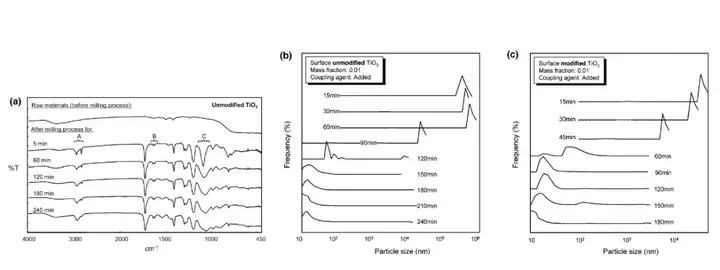
图 2 (a)经过选定的研磨时间后的二氧化钛粒子和二氧化钛- mma 悬浮液的 FTIR 光谱(A,甲氧基官能团的 C-H 拉伸;B, APTMOS 中的 CdC;C, Si-O-r 组 Si-O 拉伸)。(b)在添加偶联剂的情况下经过选定的研磨次数后,未改性和(c)经表面改性的 TiO2粒子在 MMA 中的粒径分布。在(b)和(c)中,悬浮液中的二氧化钛质
图 3 显示添加到悬浮液中的二氧化钛质量分数为 0.05 和偶联剂的表面改性二氧化钛的MMA 悬浮液的粒径分布。将悬浮液中的纳米粒子浓度增加 5 倍,团聚率大约增加了 2533倍,导致悬浮液中形成更大的团聚体(>100 μm)。破碎团聚体所需的研磨时间比具有 0.01 二氧化钛质量分数的悬浮液所需的研磨时间长几倍。 在高颗粒数浓度下,再团聚率高,使颗粒在通过珠磨机回收时重新团聚回其未研磨的大小,也就是说,在高颗粒数浓度下,再团聚率可以大于团聚破碎率 重新团聚开始发生的粒子数浓度与粒子和团聚体之间的吸引力和斥力,以及粒子和团聚体的大小和多分散性有关在这里,在悬浮液中添加偶合剂可以降低悬浮液中粒子和团聚体之间的吸引力强度,虽然需要较长的研磨时间,但珠磨能够成功地将二氧化钛团聚体分解为质量分数为 0.05 的悬浮液中的初级粒子。对悬浮液的可视化分析提供了进一步的证据,证明了珠磨分散团聚的程度。图 4 中的 a部分为经过选定的珠磨次数(顶排,1)和样品瓶中 24 h(底排,2)后的表面改性纳米二氧化钛(偶联剂,二氧化钛质量分数为 0.01)样品。经过较短的珠磨时间(小于60 min)后,样品在磨后立即不透明,24 h 后团聚的纳米粒子沉降到样品瓶底部。悬浮液由初代二氧化钛纳米粒子和纳米团聚体组成;因此,悬浮液是清晰的(悬浮液的轻微着色是由于偶联剂与纳米粒子的结合)。由于偶联剂阻止了再团聚,粒子保持纳米大小,24 小时后没有从悬浮液中沉淀出来。
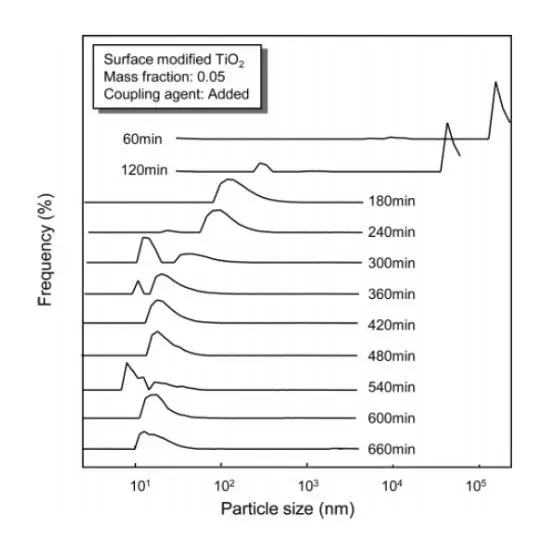
图 3 在添加偶联剂的情况下,经过选定的研磨时间后,表面改性的 TiO2 粒子在 MMA 中的粒径分布。悬浮液中二氧化钛质量分数为 0.05。
图 4 的 b 部分显示了表面改性研磨后的二氧化钛悬浮液、二氧化钛质量分数为 0.01 的二氧化钛悬浮液和偶联剂的紫外-可见透射光谱。并给出了 MMA 单体的透射光谱。MMA单体,如 PMMA,在波长大于 300 nm 时几乎有 100%的透光率。正如尺寸分布测量所预期的那样,在波长大于350 nm的二氧化钛- MMA悬浮液的透射率随着研磨时间的增加而增加,这是因为随着团聚体分散到初级粒子中,光散射量减少。然而,在 300-350 nm 的 UV 范围内,无论研磨时间多少,悬浮液几乎完全吸收。因此,分散良好的二氧化钛纳米颗粒可用于增强 PMMA 的紫外吸收性能,同时仍允许可见光的透射率。30 天后,两种悬浮液的透射光谱相同,表明分散良好的二氧化钛悬浮液非常稳定。
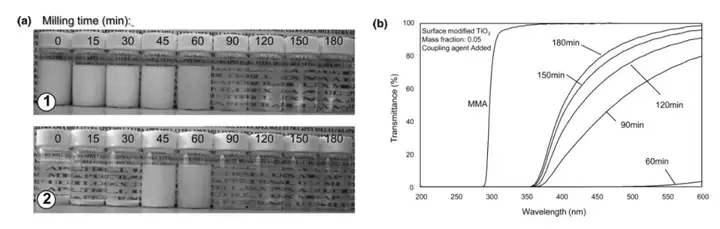
图 4 (a)添加偶联剂的表面改性二氧化钛粒子在研磨后即刻(1a)和研磨后 24 小时(2)的图像。从左到右,每个样品瓶的研磨时间分别为 0、15、30、45、60、90、120、150 和 180 分钟。(b)在选定的研磨时间后添加偶联剂的 MMA 中的表面改性二氧化钛粒子的紫外-可见透射光谱。
Titania-PMMA 纳米复合材料
由二氧化钛- MMA 悬浮液聚合形成的二氧化钛- PMMA纳米复合材料的数字图像如图 5 所示。图 5 的 a-c 部分显示的二氧化钛- PMMA 复合材料是由表面改性的二氧化钛-MMA 悬浮液聚合形成的,在没有使用珠磨的情况下,添加了偶合剂(二氧化钛和 MMA 是用砂浆和杵研磨,并与 MMA 混合),而图 5 中 d-f 部分所示的二氧化钛- PMMA 复合材料是由表面改性的二氧化钛- MMA 悬浮液与偶联剂聚合形成的,偶联剂已在珠磨机中处理过。图 5 中 a、d 部分,b、e 部分,c、f 部分的二氧化钛质量分数分别为 0.01、0.05、0.01。图 5 中零件 d、e、f 的研磨时间分别为 180、660、240 min。与单体悬浮液一样,由未研磨的悬浮液合成的二氧化钛- PMMA 复合材料是不透明的,而由研磨的悬浮液合成的复合材料是透射可见光的。由未研磨的二氧化钛- MMA 悬浮液(a,b)和(c,d)研磨的二氧化钛- MMA 悬浮液经过 660 min 合成的表面改性二氧化钛- PMMA 复合材料的 TEM 图像如图 7 所示(均添加了偶联剂)。

图 5 由(a-c)未研磨的和(d-f)研磨的二氧化钛- mma 悬浮液聚合而成的二氧化钛- pmma 复合材料图像。
图 6 中 a、c 部分悬浮液中二氧化钛的质量分数为 0.01,b、d 部分为 0.05。透射电镜图像与动态光散射法测得的二氧化钛粒径分布一致。未研磨的颗粒高度团聚,而研磨后的颗粒分散良好,从而可以合成二氧化钛- PMMA 纳米复合材料。

图 6 由(a-b)未研磨的和(c-d)研磨的二氧化钛- MMA 悬浮液聚合而成的二氧化钛- PMMA 复合材料中的二氧化钛粒子的 TEM 图像。
具有0.01 和 0.05 TiO2质量分数的 PMMA 和 PMMA-TiO2纳米复合材料(研磨 240 min 的二氧化钛- MMA 悬浮液)的重量损失与温度的曲线显示于图 7 中。
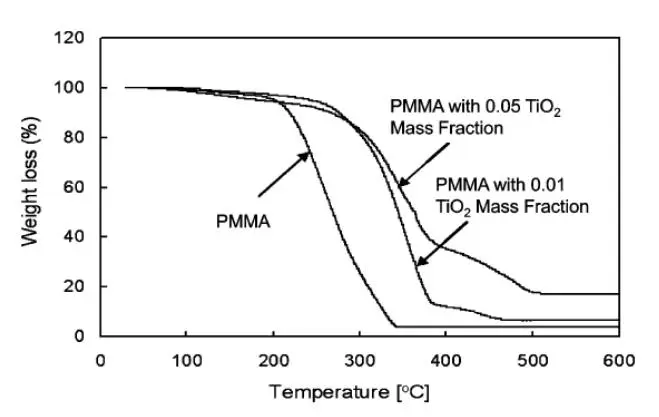
图 7 合成的 TiO2-PMMA 纳米复合材料样品和纯 PMMA 的失重与温度曲线。加热速率为 5℃/min。
通常,PMMA 的热降解分为两个步骤聚合物将首先分解成更小的聚合物段,随后,每个段将进一步分解成单体。因此,热重分析只能用于检测这一过程的第二步,因为在第一步体重损失最小。在 PMMA 中添加二氧化钛纳米颗粒来创建 PMMA- TiO2纳米复合材料提高了失重发生的温度,表明 PMMA 纳米复合材料比 PMMA 单独使用更热稳定。二氧化钛质量分数从 0.01 增加到 0.05 时,降解温度没有明显升高。这一结果可能取决于 PMMA 的分子量和 TiO2纳米粒子的大小。分解温度的升高是由于二氧化钛和 PMMA 之间的界面相互作用,PMMA 和 TiO2之间的表面接触量取决于 PMMA 的分子量和 TiO2纳米粒子的大小。
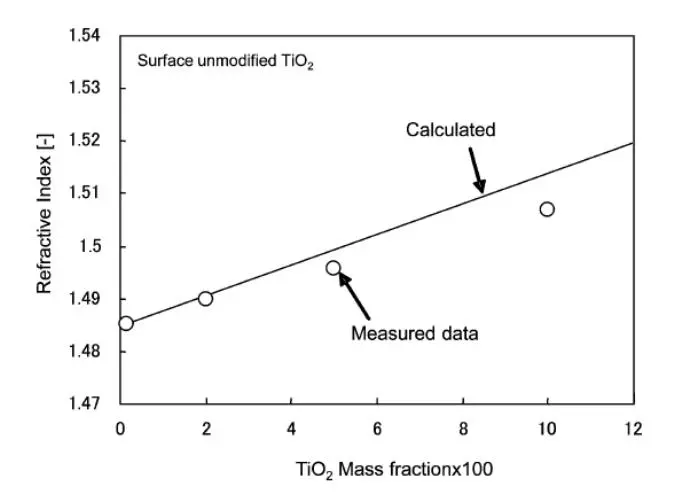
图 8 复合薄膜折射率与二氧化钛质量分数的关系
二氧化钛- PMMA 纳米复合膜的折射率显示于图 8 中随二氧化钛重量百分比的函数。纯 PMMA、PMMA 膜中 0.02、0.05、0.10 TiO2质量分数样品在 633 nm 处的折射率分别为 1.4853、1.4898、1.4958、1.5070。折射率的增加可以用 Drude 的模型来预测,
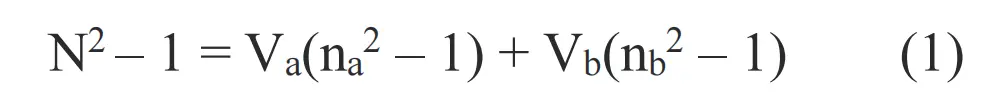
式中na、nb分别为复合聚合物 PMMA、TiO2的2.76折射率,Va、Vb分别为 PMMA、TiO2的体积分数。测量的折射率略低于模型预测的折射率,这可能与二氧化钛颗粒中存在杂质或在珠磨过程中添加了偶联剂有关。
4. 总结
采用一种新型珠磨机处理二氧化钛悬浮液和甲基丙烯酸甲酯(MMA)来破碎二氧化钛纳米颗粒团聚体。在不添加硅烷偶联剂的情况下,粒子再团聚的速度足够快,以致任何被珠磨分散的团聚体在粒子通珠磨机再循环时重新团聚。在硅烷偶联剂的作用下,在纳米颗粒质量分数高达 0.05 的悬浮液中,珠磨机能够破碎纳米颗粒团聚体。分散良好的二氧化钛纳米颗粒通过 MMA 对可见光的透射率影响不大,但增强了 MMA 的紫外吸收性能。通过在二氧化钛-MMA 悬浮液中聚合 MMA,二氧化钛- pmma 形成纳米复合材料。PMMA-二氧化钛纳米复合材料中的 PMMA 比单独的 PMMA 更热稳定。这些实验结果表明,利用新开发的珠磨机进行研磨分散是一种强大而简单的将纳米粒子分散到有机溶剂中的技术,而这正是聚合物纳米粒子复合材料合成中需要克服的主要障碍之一。
5. 参考资料
1. Schmid, G.; Talapin, D. V.; Shevchenko, E. V. In Nanoparticles: From Theory to Applications,Schmid, G., Ed.; VCH: Weinhem, Germany, 2004; p 251.
2. Haes, A. J.; Van Duyne, R. P. J. Am. Chem. Soc. 2002, 124, 10596.
3. Alivisatos, A. P. Science 1996, 271, 933.
4. Chen, S. W.; Murray, R. W.; Feldberg, S. W. J. Phys. Chem. B 1998, 102, 9898.
5. Templeton, A. C.; Pietron, J. J.; Murray, R. W.; Mulvaney, P. J. Phys. Chem. B 2000, 104,564.
6. Jin, R. C.; Cao, Y. W.; Mirkin, C. A.; Kelly, K. L.; Schatz, G. C.; Zheng, J. G. Science 2001,294, 1901.
7. Park, J. I.; Cheon, J. J. Am. Chem. Soc. 2001, 123, 5743.
8. Shevchenko, E. V.; Talapin, D. V.; Schnablegger, H.; Kornowski, A.; Festin, O.; Svedlindh, P.;Haase, M.; Weller, H. J. Am. Chem. Soc. 2003, 125, 9090.
9. Li, Y.; Boone, E.; El-Sayed, M. A. Langmuir 2002, 18, 4921.
10. Tsunoyama, H.; Sakurai, H.; Ichikuni, N.; Negishi, Y.; Tsukuda, T. Langmuir 2004, 20,11293.
11. Oberdisse, J. Soft Matter 2006, 2, 29.
12. Zhao, H. X.; Li, R. K. Y. J. Polym. Sci., Part B: Polym. Phys. 2005, 43, 3652.
13. Kuo, M. C.; Tsai, C. M.; Huang, J. C.; Chen, M. Mater. Chem. Phys. 2005, 90, 185.
14. Cole, D. H.; Shull, K. R.; Baldo, P.; Rehn, L. Macromolecules 1999, 32, 771.
15. Ash, B. J.; Siegel, R. W.; Schadler, L. S. J. Polym. Sci., Part B: Polym. Phys. 2004, 42, 4371.
16. Carter, S. A.; Scott, J. C.; Brock, P. J. Appl. Phys. Lett. 1997, 71, 1145.
17. Benaissa, M.; JoseYacaman, M.; Xiao, T. D.; Strutt, P. R. Appl. Phys. Lett. 1997, 71, 3685.
18. Schmidt, G.; Malwitz, M. M. Curr. Opin. Colloid Interface Sci. 2003, 8, 103.
19. Pramanik, P. Bull. Mater. Sci. 1995, 18, 819.
20. Sambhy, V.; MacBride, M. M.; Peterson, B. R.; Sen, A. J. Am. Chem. Soc. 2006, 128, 9798.
21. Yang, M. J.; Dan, Y. J. Appl. Polym. Sci. 2006, 101, 4056.
22. Pavel, F. M.; Mackay, R. A. Langmuir 2000, 16, 8568.
23. Raula, J.; Shan, J.; Nuopponen, M.; Niskanen, A.; Jiang, H.; Kauppinen, E. I.; Tenhu, H.Langmuir 2003, 19, 3499.
24. Kammler, H. K.; Madler, L.; Pratsinis, S. E. Chem. Eng. Technol. 2001, 24, 583.
25. Artelt, C.; Schmid, H. J.; Peukert, W. J. Aerosol Sci. 2005, 36, 147.
26. Israelachvili, J. Intermolecular and Surface Forces; Academic Press: London, 2000.
27. Muller, F.; Peukert, W.; Polke, R.; Stenger, F. Int. J. Miner. Process. 2004, 74, S31.
28. Inkyo, M.; Tahara, T.; Iwaki, T.; Iskandar, F.; Hogan, C. J.; Okuyama, K. J. Colloids Interface Sci. 2006, 304, 535.
29. Inkyo, M.; Tahara, T. J. Soc. Powder Technol., Japan 2004, 41, 578.
30. Sommer, M.; Stenger, F.; Peukert, W.; Wagner, N. J. Chem. Eng. Sci. 2006, 61, 135.
31. Stenger, F.; Mende, S.; Schwedes, J.; Peukert, W. Chem. Eng. Sci. 2005, 60, 4557.
32. Tsantilis, S.; Pratsinis, S. E. Langmuir 2004, 20, 5933.
33. Friedlander, S. K., Smoke, Dust, and Haze: Fundamentals of Aerosol Dynamics, 2nd ed.;Oxford University Press: New York, Oxford, 2000.
34. Yang, F.; Nelson, G. L. J. Appl. Polym. Sci. 2004, 91, 3844.